Imran Ahamed, Kanchan Ulman, Nicola Seriani, Ralph Gebauer, Arti Kashyap
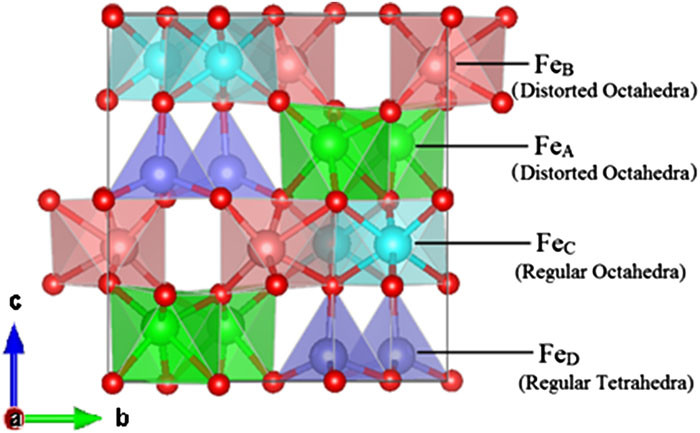
The metastable iron oxide ϵ-Fe2O3 is rare but known for its magnetoelectric properties. While the more common alpha phase has been recognized for a long time as a suitable material for photoelectrochemical cells, its use is limited because of the electron-hole recombination problem when exposed to light. The indirect bandgap of the epsilon phase with its spontaneous polarization may offer a better potential for the application in photoelectrochemistry. Here, we report a detailed study of the electronic and structural features of the epsilon phase of iron oxide, its stability in thin films, and possible water dissociation reactions. Our studies are performed using density functional theory with a Hubbard-U correction. We observe that the stable ϵ-Fe2O3 surfaces favor the dissociation of water. The average difference in the energies of the states when water is adsorbed and when it is dissociated is roughly found to be −0.40 eV. Our results compare with the available experimental results where the epsilon phase is reported to be more efficient for the release of hydrogen from renewable oxygenates when exposed to sunlight.